Epilepsy - Role of MRI
Laurens De Cocker, Felice D'Arco and Philippe Demaerel and Robin Smithuis
Publicationdate
In many patients with epilepsy antiepileptic drug treatment is unable to control the seizures.
Using a dedicated MRI-protocol, it is possible to detect an epileptogenic lesion in 80 percent of these patients.
Resection of these lesions can lead to seizure freedom in many patients.
We will discuss the MRI protocol and the typical findings in the most common epilepsy-associated diseases.
Introduction
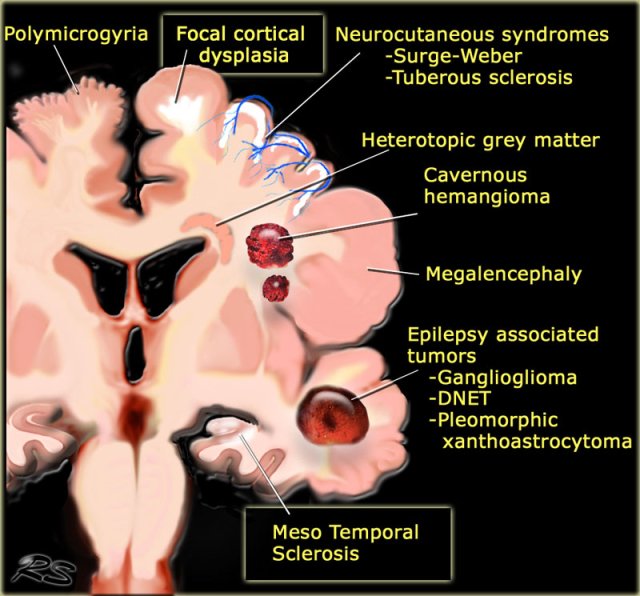
Common causes of Epilepsy
The illustration summarizes the most common causes of seizures in patients with medically uncontrollable epilepsy.
Some of these lesions are readily identifiable.
Meso temporal sclerosis and focal cortical dysplasia are the most common causes and can only be depicted with a dedicated protocol.

The table also summarizes epileptogenic lesions that are detected in patients with uncontrollable seizures.
Mesial temporal sclerosis is the most common cause of intractable epilepsy.
In medication refractory epilepsia the most common location of the epilectogenic lesion is temporal lobe (60%), frontal lobe (20%) and parietal lobe (10%), periventricular (5%) and occipital (5%).
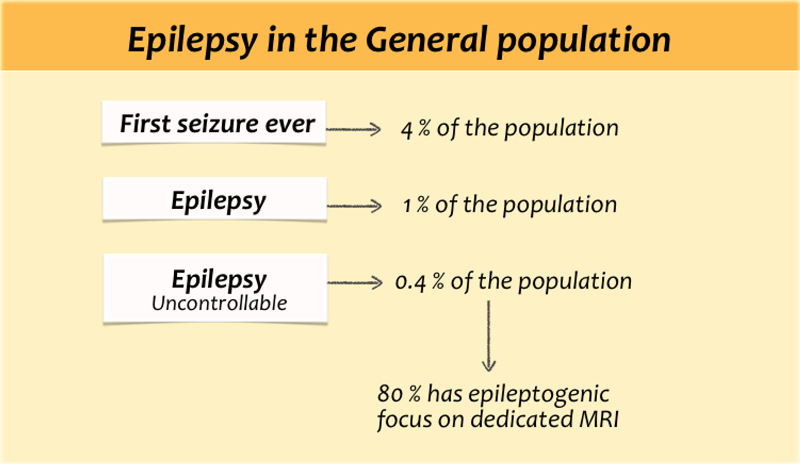
Seizures and Epilepsy
Seizures are common. About 4 percent of all people will have at least one seizure during their lifetime.
In patients with a first ever seizure imaging will mostly show no brain-abnormalities, because the seizure is provoked by fever, drugs, dehydration or sleep deprivation.
The term epilepsy is used, when there are recurrent unprovoked seizures.
About 60 percent of patients with epilepsy can be controlled with antiepileptic drugs.
Most patients with uncontrollable seizures have complex partial seizures.

Partial seizures - also called focal seizures - are seizures which affect only a part of the brain at onset. They usually start in the temporal lobe.
In simple partial seizures the person remains conscious.
A simple partial seizure can be a precursor to a larger seizure and then it is called an aura.
A complex partial seizure affects a larger part of the hemisphere and the person may lose consciousness.
If a partial seizure spreads from one hemisphere to the other this will give rise to a secondarily generalised seizure.
The person will become unconscious and may have a tonic clonic seizure.

MRI epilepsy protocol
The table shows a dedicated epilepsy protocol.
Some will also use Inversion Recovery and not use contrast on a routine base.
T1WI
Superior for cortical thickness and the interface between grey and white matter.
On T1WI look for grey matter occuring in an aberrant location as in gray matter heterotopia.
FLAIR
Look very carefully for cortical and subcortical hyperintensities on the FLAIR, which can be very subtle.
Since FLAIR may show false-positive results due to artefacts, the abnormalities should be confirmed on T2WI.
T2* or SWI
Helpful when searching for haemoglobin breakdown products as in posttraumatic changes and cavernomas, or to look for calcifications in tuberous sclerosis, Sturge-Weber, cavernomas and gangliogliomas.
Mesial temporal sclerosis
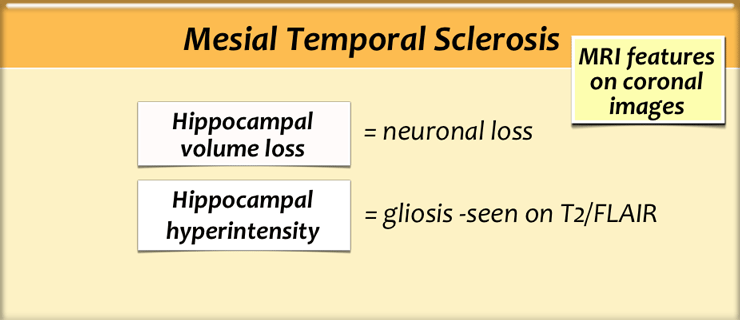
Mesial temporal sclerosis (MTS) is a specific pattern of hippocampal neuronal loss accompanied by gliosis and atrophy.
The etiology is unknown, but there is a relationship between MTS and prolonged febrile seizures earlier in life, complicated delivery and developmental processes.
In 15% of patients another developmetal abnormality can be found, mostly focal cortical dysplasia.
This is called dual pathology.
MTS is the most common cause of partial complex epilepsy in adults and is also the most common etiology in young adult patients undergoing surgery.
Surgical removal of visible MRI changes associated with unilateral mesial temporal sclerosis leads to seizure freedom in up to 80% of cases.
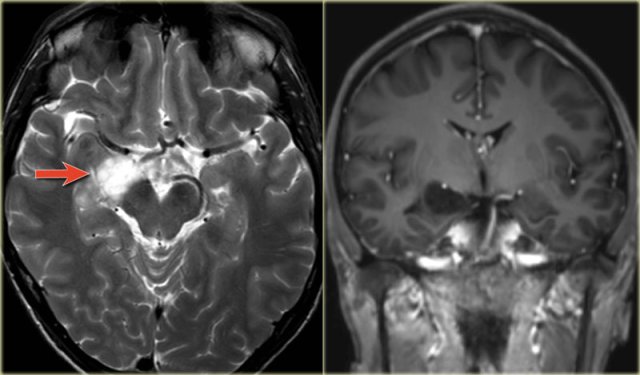
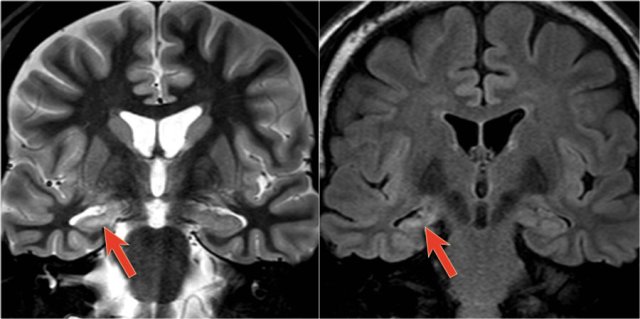 Right-sided mesial temporal sclerosis
Right-sided mesial temporal sclerosis
Coronal T2W and FLAIR images are the most sensitive for detecting MTS.
On axial slices mesial temporal sclerosis is commonly overlooked.
Bilateral mesial temporal sclerosis is difficult to detect due to the lack of comparison with the unaffected contralateral hippocampus.
The coronal T2WI and FLAIR images show right-sided mesial temporal sclerosis.
Notice the volume loss, which indicates atrophy and causes secondary enlargement of the temporal horn of the lateral ventricle.
The high signal in the hippocamous reflects gliosis.
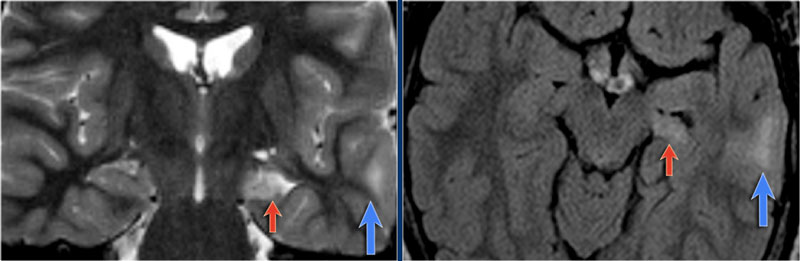 Dual pathology: MTS and focal cortical dysplasia
Dual pathology: MTS and focal cortical dysplasia
Mesial temporal sclerosis may occur in association with other pathology, especially focal cortical dysplasia.
This is called dual pathology.
The images show mesial temporal sclerosis with a hyperintense and shrunken hippocampus (red arrows), and secondary enlargement of the left temporal horn of the left laterale ventricle.
Also notice associated subcortical hyperintensity in the left temporal lobe indicating focal cortical dysplasia.
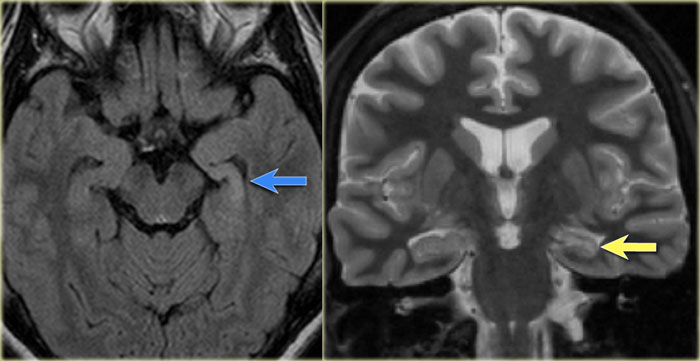 Left mesial temporal sclerosis. Subtle gliosis of left hippocampus (blue arrow) and atrophy (yellow arrow).
Left mesial temporal sclerosis. Subtle gliosis of left hippocampus (blue arrow) and atrophy (yellow arrow).
35-year-old patient with refractory temporal lobe epilepsy.
MR shows subtle hyperintensity of the left hippocampus on the axial FLAIR (blue arrow) and atrophy of the left hippocampus on coronal images (yellow arrow).
 Left mesial temporal sclerosis treated with amygdalo-hippocampectomy
Left mesial temporal sclerosis treated with amygdalo-hippocampectomy
The patient was succesfully treated with amygdalo-hippocampectomy on the left.
Differential of hippocampal hyperintensity
Hippocampal hyperintensity on T2WI or FLAIR images with volume loss is diagnostic for mesial temporal sclerosis in the appropriate clinical setting.
Hippocampal hyperintensity without volume loss is seen in:
- Status epilepticus
- Low grade tumors (astrocytoma, DNET)
- Encephalitis
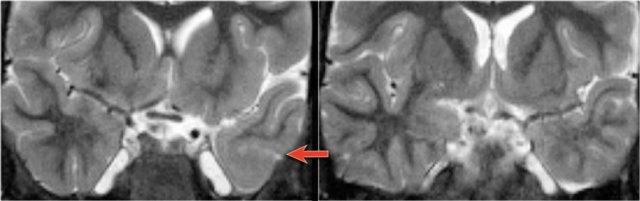
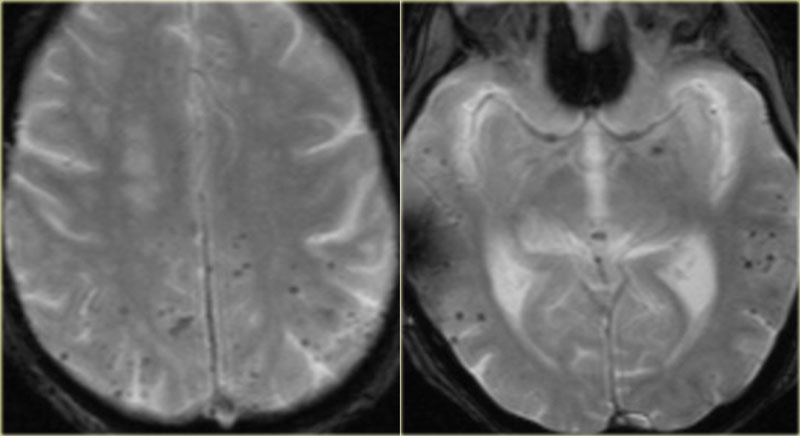
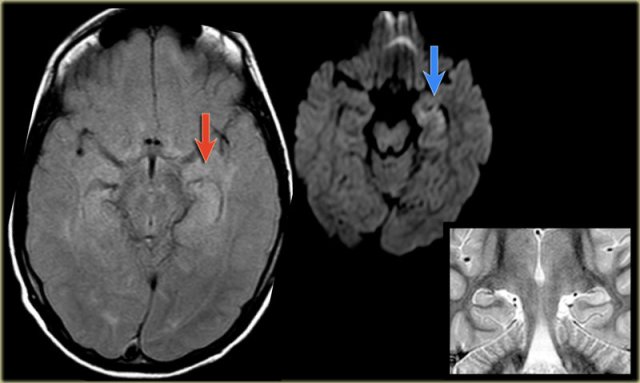 Status epilepticus. Axial FLAIR, axial DWI and coronal T2WI.
Status epilepticus. Axial FLAIR, axial DWI and coronal T2WI.
Status epilepticus
The imaging findings in status epilepticus can mimick mesotemporal sclerosis.
In status epilepticus a hyperintense hippocampus can be seen, but there is swelling and no atrophy.
Axial FLAIR, axial DWI and coronal T2WI demonstrate a hyperintense hippocampus with a slightly compressed temporal horn of the lateral ventricle consistent with hippocampal edema.
DWI shows diffusion restriction due to cytotoxic edema in the acute stage of the status epilepticus.
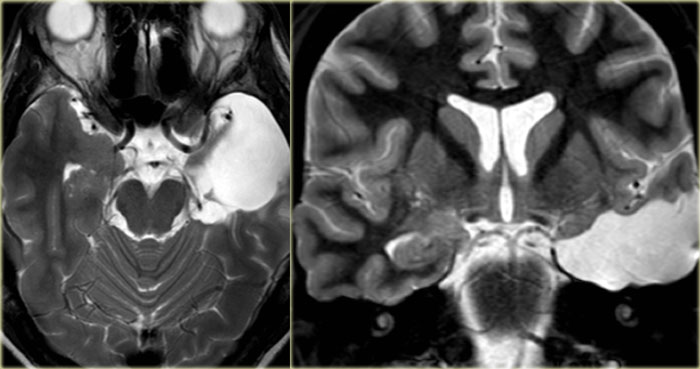
 Hyperintense hippocampus due to a DNET
Hyperintense hippocampus due to a DNET
DNET mimicking mesial temporal sclerosis
Axial T2WI shows hyperintense, but enlarged hippocampus with a bubbly appearance.
This is typical for a DNET or dysembryoplastic neuroepithelial tumor, which we will discuss in a moment.
The coronal contrast-enhanced T1WI shows an enlarged hippocampus without uptake of contrast medium.
Focal Cortical Dysplasia

key findings
- Subcortical white matter hyperintensities
- Blurred grey-white matter interface
Focal cortical dysplasia is a congenital abnormality where the neurons fail to migrate in the proper formation in utero.
MRI findings may be very subtle or may even be negative, therefore a high index of suspicion is mandatory!
The most common findings are cortical or subcortical hyperintensities especially seen on FLAIR-images.
These are often found at the bottom of a deep sulcus.
Another finding is a blurred interface between grey and white matter, because the white matter looks a little bit like gray matter because it contains neurons that did not reach the cortex.
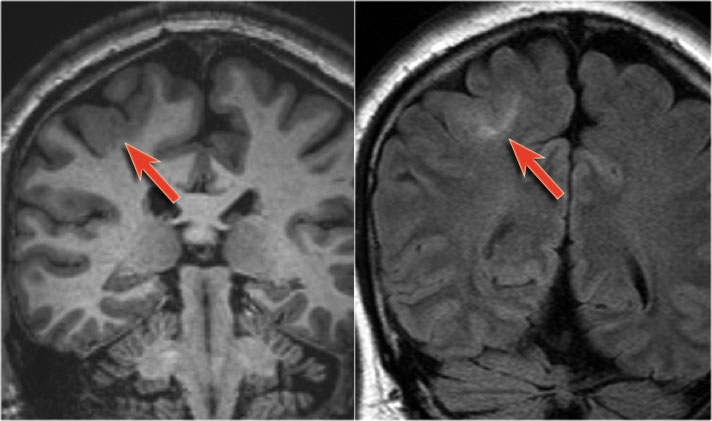 Focal cortical dysplasia - coronal T1WI and FLAIR
Focal cortical dysplasia - coronal T1WI and FLAIR
The images show typical focal cortical dysplasia.
There is cortical thickening and blurring of the grey/white matter junction on T1WI (left).
The FLAIR image on the right shows the subcortical hyperintensity.
 Dual pathology: MTS and focal cortical dysplasia
Dual pathology: MTS and focal cortical dysplasia
The images demonstrate cortical and subcortical signal abnormalities on T2WI and FLAIR in the left temporal lobe indicating focal cortical dysplasia.
Notice associated T2/FLAIR hyperintense and shrunken hippocampus as a result of mesial temporal sclerosis, i.e. dual pathology.
 Focal cortical dysplasia. Coronal T2WI
Focal cortical dysplasia. Coronal T2WI
Another case of focal cortical dysplasia.
Notice the hypoplastic left temporal lobe with cortical thickening (arrow) and atrophy of the white matter.
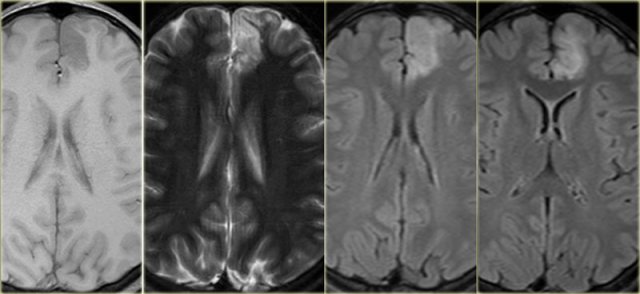 Focal cortical dysplasia. T1WI, T2WI and FLAIR.
Focal cortical dysplasia. T1WI, T2WI and FLAIR.
Axial T1WI, T2WI and FLAIR-images of a 15 year old boy with epilepsy.
Notice thickening and hyperintensity of the cortex of the left superior frontal gyrus.
The FLAIR-images also show high signal in the subcortical white matter.
These findings are typical for focal cortical dysplasia.
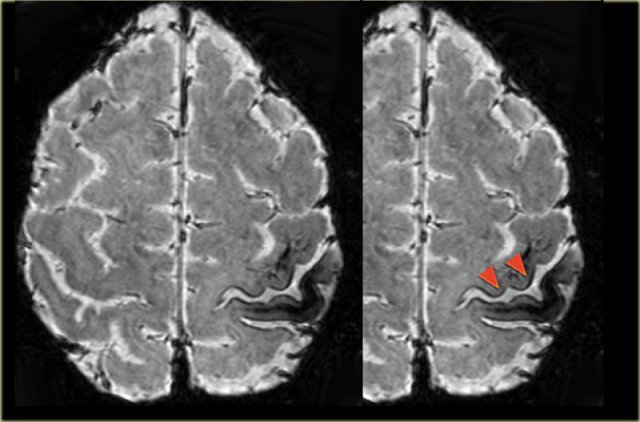
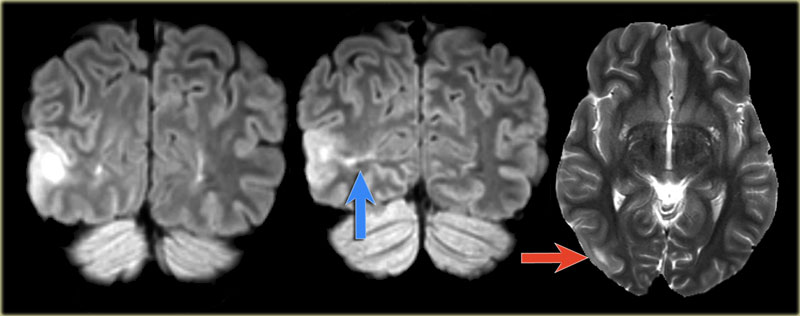 Focal cortical dysplasia in the right occipital lobe with transmantle sign
Focal cortical dysplasia in the right occipital lobe with transmantle sign
Transmantle sign
Sometimes the hyperintensity is seen extending from the subcortical area to the margin of the ventricle.
This is called the transmantle sign.
This finding represents the arrested neuronal migration.
Images of a 27-year-old male with refractory occipital lobe epilepsy.
Coronal FLAIR and axial T2WI show T2-hyperintense cortical thickening and high signal in cortex and subcortical region.
Notice subcortical hyperintensity extending to the right ventricle indicating transmantle sign (blue arrow).
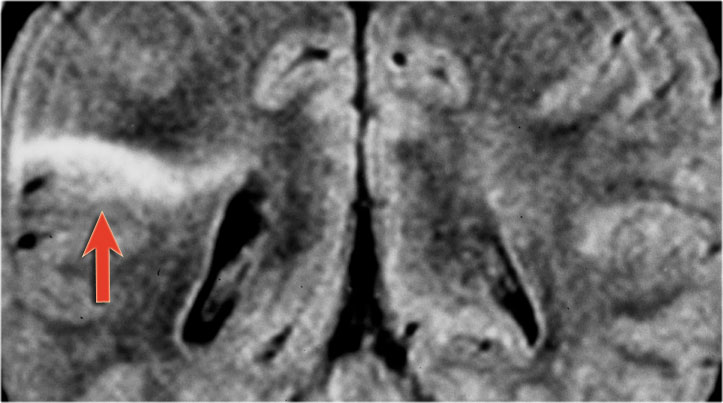 Focal cortical dysplasia - Transmantle sign.
Focal cortical dysplasia - Transmantle sign.
Transmantle sign seen in another patient with focal cortical dysplasia.
Cortical and glial scars - Ulegyria
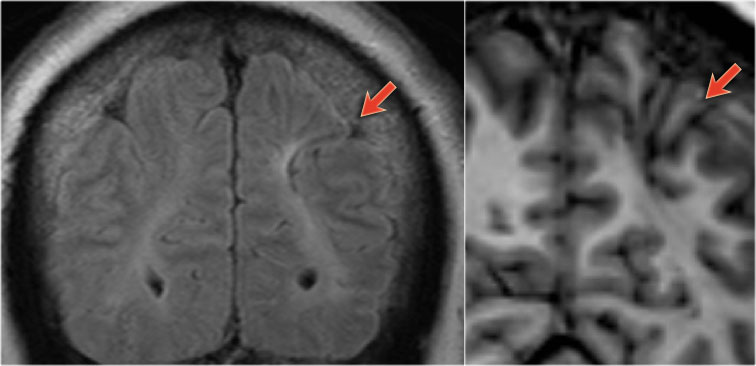 54-year-old patient with a history of perinatal asphyxia and longstanding refractory partial epilepsia. Left parietal scar in the parasagittal watershed area resulting in a shrunken cortex.
54-year-old patient with a history of perinatal asphyxia and longstanding refractory partial epilepsia. Left parietal scar in the parasagittal watershed area resulting in a shrunken cortex.
Cortical and glial scars usually result from meningitis or birth injury.
Ulegyria is a specific type of scar.
It is defined as cerebral cortex scarring due to perinatal ischemia.
Ulegyria typically affects full term infants.
In these infants there is greater perfusion to the apex of the gyri than to the cortex at the depth of the sulci.
The resulting pattern is that of a shrunken cortex in which the deep portions of the gyri are more shrunken than the superficial portions, leaving pedunculated gyri on long stalks with a mushroom appearance.
Ulegyria must be differentiated from microgyria.
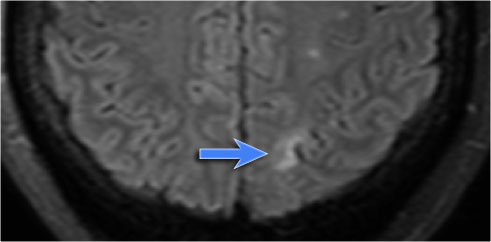 Ulegyria
Ulegyria
MR will shows tissue loss and gliosis underneath a shrunken cortex.
The shrunken cortex is best appreciated on a 3D-T1WI because of its high resolution and the superior delineation of the cortex, while FLAIR will show the hyperintensity associated with the gliosis.
Therefore always use the FLAIR-sequence to search for hyperintensities in an epileptic patient and subsequently correlate these findings with the cerebral cortex in the affected area on high resolution T1WI.
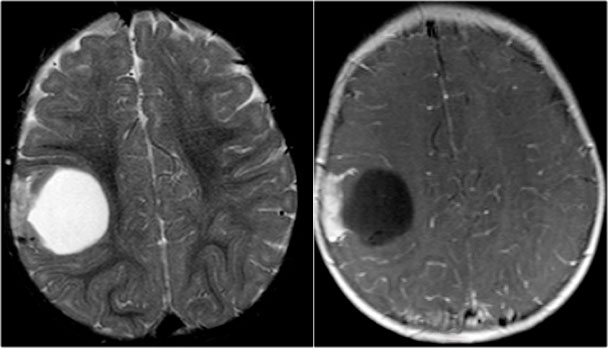
Cavernoma

Cavernoma is also known as cavernous malformation or cavernous angioma.
It is a benign low flow vascular malformation with a tendency to bleed.
75 percent occur as solitary sporadic lesions and 10-30 percent occur as multiple lesions.
Cavernomas consist of locules of variable size that contain blood products in different stages of evolution which produces a popcorn appearance.
A complete hemosiderin rim surrounds the lesion, but not when there is a recent bleeding.
Unenhanced CT may show a hyperdense nodule or calcification, but in 50% of cases cavernomas will be occult on CT.
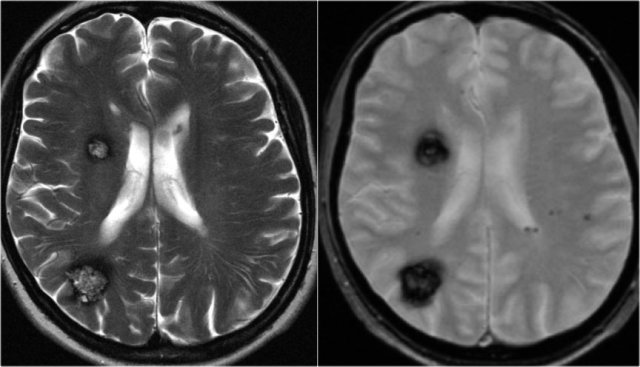
T2WI and T2* gradient echo show multiple cavernomas.
Notice the popcorn appearance with peripheral rim of hemosiderin on the T2WI.
The lesions are almost completely black on the gradient echo due to blooming artefacts.
T2* and susceptibility weighted imaging (SWI) markedly increase the sensitivity of MRI to detect small cavernomas.
The five black dots in the left cerebral hemisphere on the T2* are also cavernomas and are not visible on the T2WI.
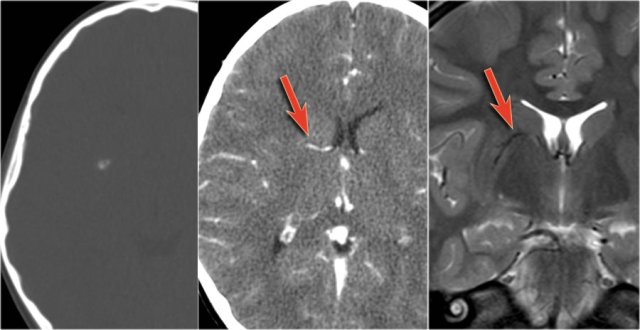 Cavernoma with associated DVA (arrow).
Cavernoma with associated DVA (arrow).
Cavernomas are associated with developmental venous anomalies (DVA's).
The unenhanced CT shows a small calcification in the right lentiform nucleus.
Enhanced CT shows a venous anomaly draining the cavernoma into the right internal cerebral vein.
Coronal T2WI shows the venous anomaly as a curvilinear flow void.
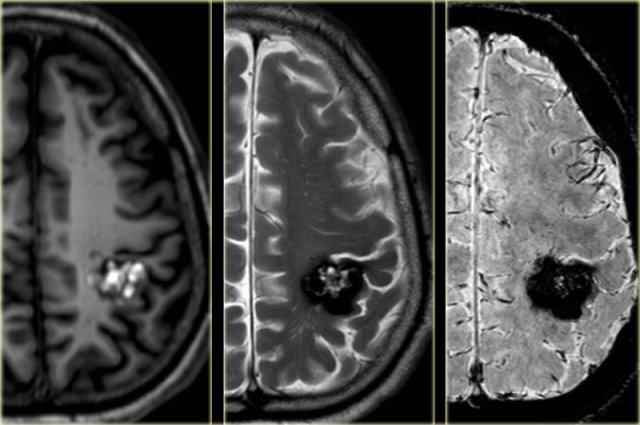
Cavernoma in the postcentral gyrus on T1WI, T2WI and SWI.
Notice popcorn appeance and blooming artefact.
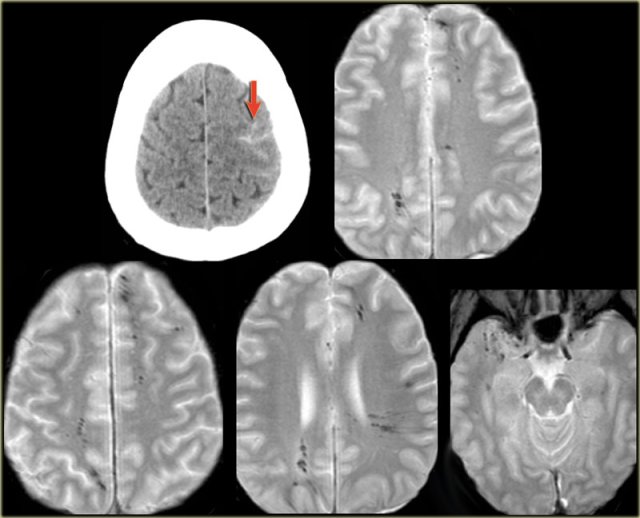
Same patient.
Notice the hemosiderin coating of the precentral gyrus consistent with superficial siderosis due to prior hemorrhage of the cavernoma (red arrowheads).
 CAA - Multifocal subcortical black dots in a older patient.
CAA - Multifocal subcortical black dots in a older patient.
Differential diagnosis of microbleeds
In patients with multiple small black dots the differential diagnosis is:
- Cavernomas
- Cerebral amyloid angiopathy (CAA)
Asymmetric microbleeds in peripheral location seen in normotensive older person with lobar hemorrhage. CAA is commonly seen in demented patients. - Hypertensive microhemorrhages
Microbleeds in hypertensive patients younger than patients with CAA - Diffuse axonal injury (DAI)
Posttraumatic hemorrhages in corpus callosum, subcortical white matter and brainstem.
 Diffuse axonal injury
Diffuse axonal injury
Diffuse axonal injury (DAI)
A 46 year old biker presented with seizures after being hit by a car.
CT-image shows only minimal subarachnoidal hemorrhage (arrow).
MRI was performed several weeks after the injury because of a change in personality.
T2*-images show multiple hemosiderin depositions at the interface between grey and white matter, consistent with diffuse axonal injury (DAI).
Notice that the location of the microbleeds is different from the peripheral located CAA-bleeds.
Epilepsy associated tumours

All brain tumors may present with epilepsy, but there are some typically epilepsy associated tumors.
These tumours share the following characteristics:
- They arise in a cortical location.
- Often located in the temporal lobe.
- Closely related to developmental malformations.
- Typically seen in adolescents and young adults.
- Characterized by a benign behaviour, a slow growth, a sharp delineation and usually show absence of edema.
- Show signs of chronicity, such as bone remodeling and scalloping of the adjacent skull.
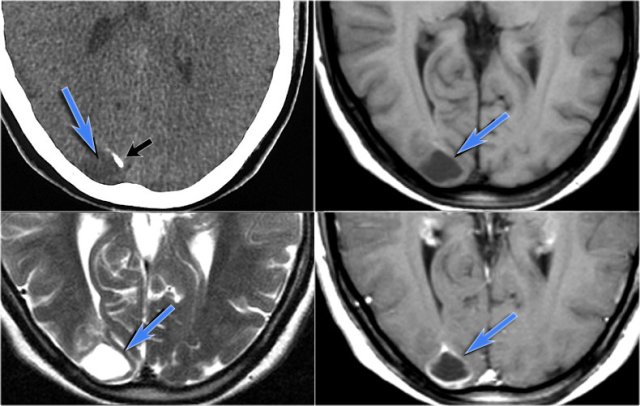 Ganglioglioma in the right occipital lobe presenting as a cystic mass with rim enhancement. Notice calcification on CT.
Ganglioglioma in the right occipital lobe presenting as a cystic mass with rim enhancement. Notice calcification on CT.
Ganglioglioma
key findings
- Typically presents as cyst with enhancing mural nodule, but may be entirely solid
- Calcification in up to 50%
Ganglioglioma is the most common tumor associated with temporal lobe epilepsy.
Calcification is common in ganglioglioma and is an important distinguishing factor from DNET and pleomorphic xanthoastrocytoma.
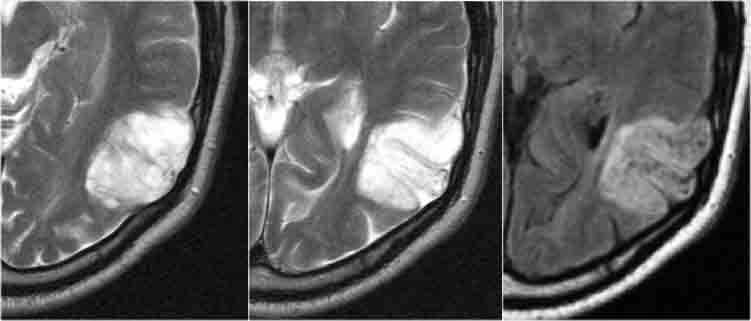
 Ganglioglioma. T2WI and CE-T1WI
Ganglioglioma. T2WI and CE-T1WI
Ganglioglioma in a young child. Note large cyst with enhancement of mural solid tissue.
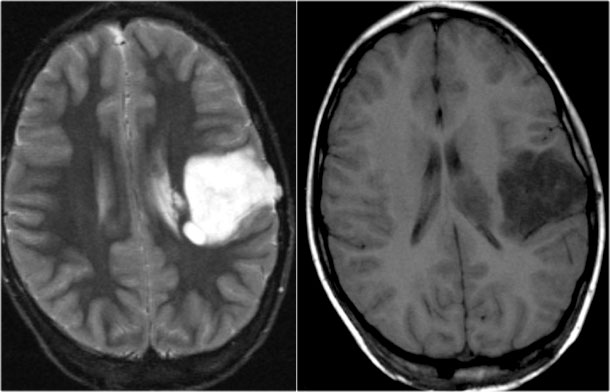
 Ganglioglioma. T2WI and CE-T1WI
Ganglioglioma. T2WI and CE-T1WI
Small cystic ganglioglioma with a small enhancing nodule.
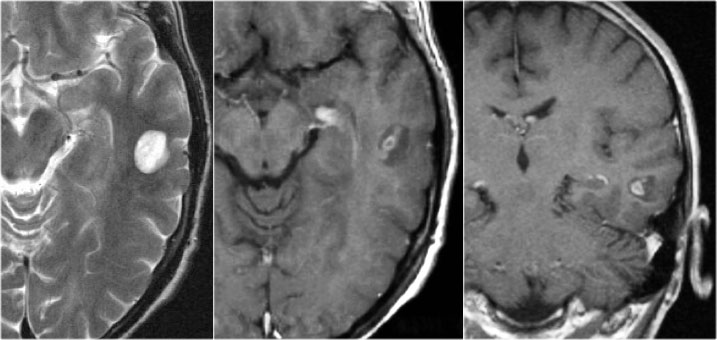
 DNET: T2WI and FLAIR show characteristic bubbly appearance and swelling of affected gyri. Notice scalopping of the skull due to slow growth of the lesion.
DNET: T2WI and FLAIR show characteristic bubbly appearance and swelling of affected gyri. Notice scalopping of the skull due to slow growth of the lesion.
DNET
key findings
- Swollen gyrus
- Bubbly cystic appearance
- May be wedge shaped and point towards the ventricle
- Usually no or only little enhancement
- Associated with focal cortical dysplasia
DNET in typical cases present as a bubbly mass which expands the affected gyri.
The bubbly cystic appearance is seen as small cyst-like intratumoral structures that are very hyperintense on T2WI.
 DNET. T2WI and T1WI
DNET. T2WI and T1WI
DNET in an 11-year old boy presenting with refractory partial seizures.
The tumor shows a characteristic bubby appearance and there is subtle scalopping of the skull.
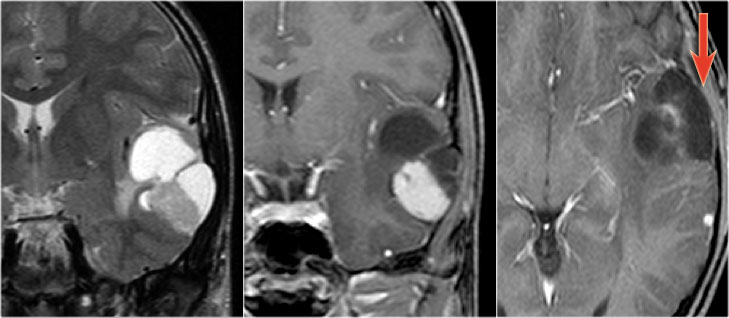 Pleomorphic xanthoastrocytoma on coronal T2WI and a coronal and axial CE-T1WI.Notice characteristic meningocerebral enhancement (arrow)
Pleomorphic xanthoastrocytoma on coronal T2WI and a coronal and axial CE-T1WI.Notice characteristic meningocerebral enhancement (arrow)
Pleomorphic xanthoastrocytoma
key findings
- Supratentorial cyst with enhancing mural nodule which abuts the peripheral meninges
- Meningeal contrast enhancement
- Peritumoral edema
Pleomorphic xanthoastrocytoma (PXA) is a rare cause of temporal lobe epilepsy.
Peritumoral edema may be seen in PXA, while it is not a feature of either ganglioglioma or DNET.
Thickening and enhancement of the adjacent leptomeninges is highly characteristic but is not always present.
When meningeal involvement is not present, than a pleiomorphic xanthoastrocytoma is indistinguishable from a ganglioglioma.
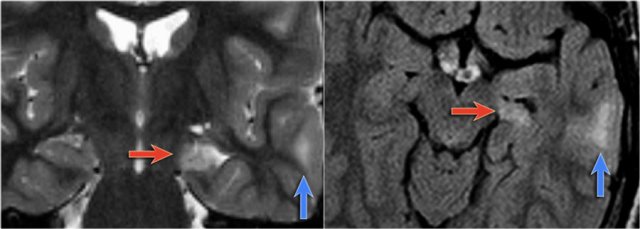
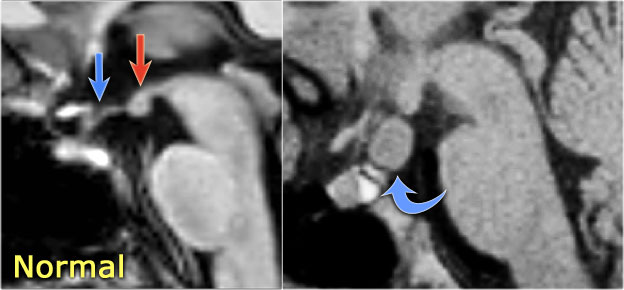 LEFT: Normal infundibular recess of the third ventricle (blue arrow) , mamillary bodies (red arrow)RIGHT: Tuber cinereum hamartoma (curved arrow)
LEFT: Normal infundibular recess of the third ventricle (blue arrow) , mamillary bodies (red arrow)RIGHT: Tuber cinereum hamartoma (curved arrow)
Hypothalamic hamartoma
key findings
- Non-enhancing enlargement of the tuber cinereum of the hypothalamus
Hypothalamic hamartoma is also known as diencephalic or tuber cinereum hamartoma.
It represents nonneoplastic congenital grey matter heterotopia in the region of tuber cinereum of the hypothalamus.
It is seen in infants presenting with seizures and precocious puberty.
Hemimegalencephaly
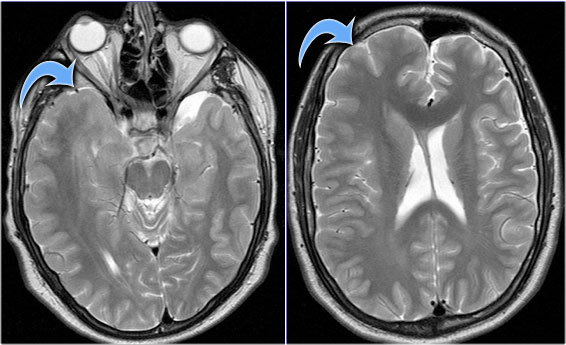 Hemimegalencephaly. (Courtesy of Alessandra D'Amico)
Hemimegalencephaly. (Courtesy of Alessandra D'Amico)
key findings
- Enlarged hemisphere with ipsilateral ventriculomegaly
T2WI shows right hemimegalencephaly.
Notice the asymmetric skull and slightly enlarged lateral ventricle.
Hemimegalencephaly is the only condition in which an increase in parenchymal volume is associated with an increase in ipsilateral ventricular volume.
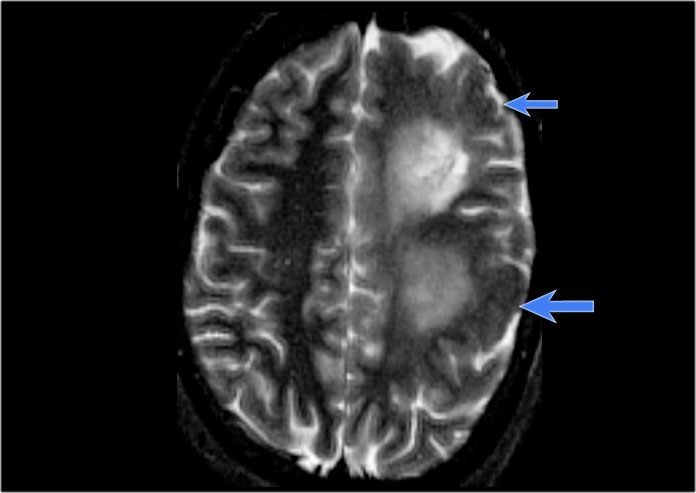 Left hemimegalencephaly with diffuse cortical dysplasia involving fronto-parietal regions (blue arrows) and diffuse white matter T2 hyperintensity
Left hemimegalencephaly with diffuse cortical dysplasia involving fronto-parietal regions (blue arrows) and diffuse white matter T2 hyperintensity
Hemimegalencephaly is a rare disease characterized by hamartomatous growth of one cerebral hemisphere or part of it.
Patients present with early seizures, macrocrania and severe developmental delay with contralateral hemiparesis.
The thickened cortex may show a wide spectrum of abnormalities, such as lissencephaly, pachygyria or polymicrogyria.
In the late stage, the involved hemisphere may atrophy due to constant seizure acitivity.
Most of the affected children die in the first years of life because of status epilepticus.
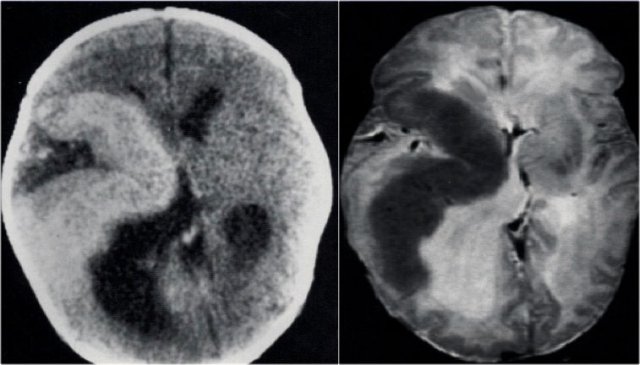 Hemimegalencephaly. Courtesy of Alessandra D'Amico
Hemimegalencephaly. Courtesy of Alessandra D'Amico
CT and T2WI in a patient with a right hemimegalencephaly.
There is dysplastic thick cortex and ventricular dilatation on the affected side.
9-y-old girl with refractory nocturnal epilepsy.
MRI shows overgrowth of the left cerebral hemisphere.
T1WI shows heterotopic gray matter lining the left lateral ventricle (blue arrow).
In hemimegalencephaly it is important to exclude contralateral abnormalities, as these form a contraindication to hemispherectomy.
Rasmussen's Encephalitis
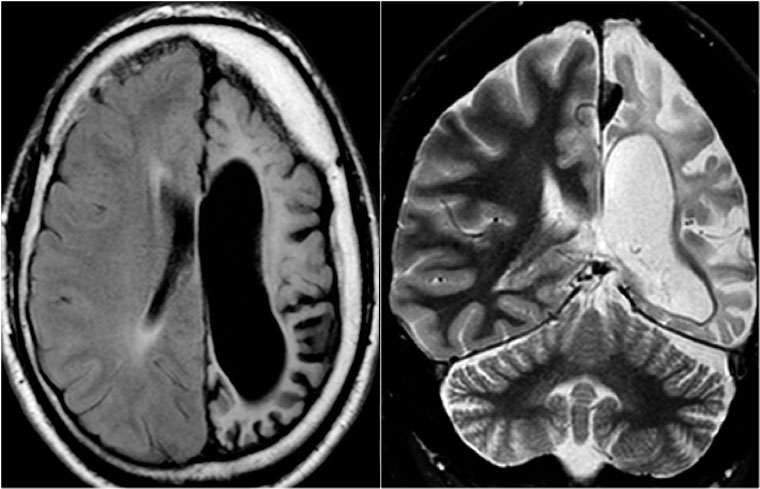 Rasmussen's encephalitis. Axial FLAIR and coronal T2WI show atrophy of the left cerebral hemisphere with enlargement of the lateral ventricle.
Rasmussen's encephalitis. Axial FLAIR and coronal T2WI show atrophy of the left cerebral hemisphere with enlargement of the lateral ventricle.
key findings
- Intractable seizure activity in children
- Progressive atrophy of the involved hemispere
- Small hemisphere with large ventricle
Rasmussen's encephalitis is a progressive hemispheric atrophy of unknown origin.
Patient develop an increasing frequency of seizures and progressive hemiplegia.
Notice that, opposed to hemimegalencephaly, the smaller hemisphere is the site of abnormality, and the lateral ventricle is larger in the smaller hemisphere.
Tuberous Sclerosis
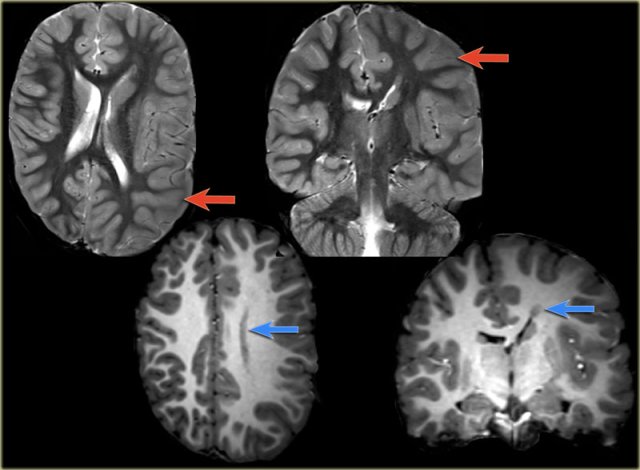
 Axial T2w shows multiple tubers and white matter abnormalities (fig. a: arrows) and subependymal nodules
Axial T2w shows multiple tubers and white matter abnormalities (fig. a: arrows) and subependymal nodules
key findings in the brain
- Cortical hamartomas
- Subependymal tubers
- Subependymal giant cell astrocytoma
- White matter abnormalities
Tuberous sclerosis or Bourneville's disease is an inherited condition characterized by the presence of hamartomas in many organs including angiomyolipoma of the kidney, cardiac rhabdomyoma and cortical and subependymal tubers in the brain.
Some patients have lymphangioleiomatosis, a cystic lung disease seen in women.
The classic clinical triad is focal epilepsy, adenoma sebaceum and mental retardation (mnemonic: fits, zits and nitwits).
The cortical hamartomas are called tubers and are similar to cortical dysplasia.
Subependymal nodules are small lesions protruding into the lateral ventricles. Sometimes they are calcified.
Seizure surgery in TSC is contemplated if a particular tuber can be implicated in seizure activity, or if a subependymal giant cell astrocytomas obstructs the foramen of Monro causing hydrocephalus.
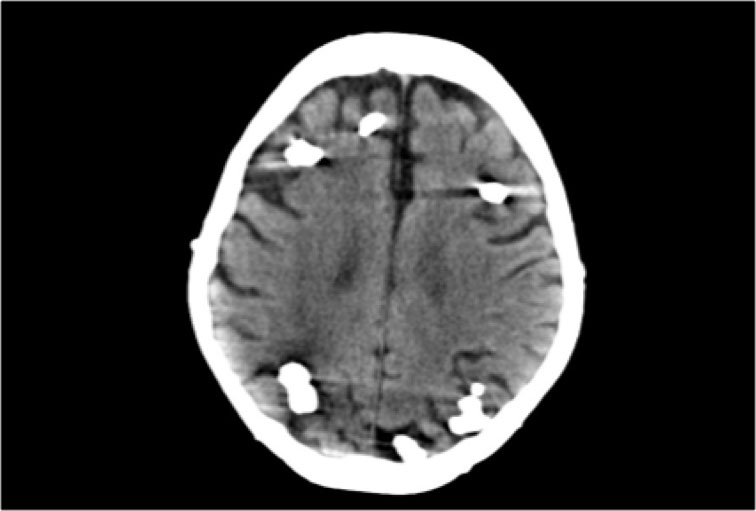
CT of a patient with Tuberous Sclerosis shows multiple cortical and subcortical calcifications.
 Tuberous Sclerosis - courtesy of the American Journal of Neuroradiology.
Tuberous Sclerosis - courtesy of the American Journal of Neuroradiology.
CT and MRI in a patient with Tuberous Sclerosis.
There are multiple cortcal and subependymal nodules.
The CT shows that most of the lesions are calcified.
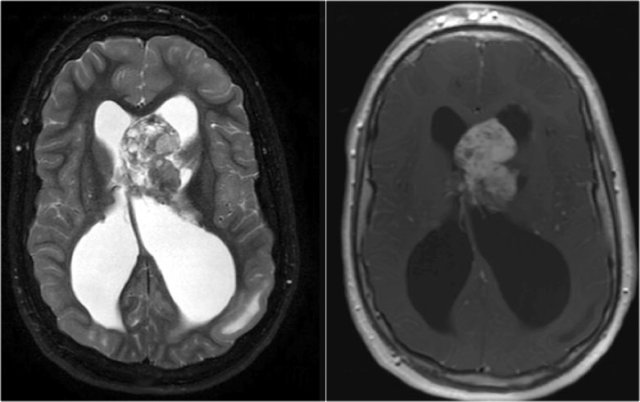
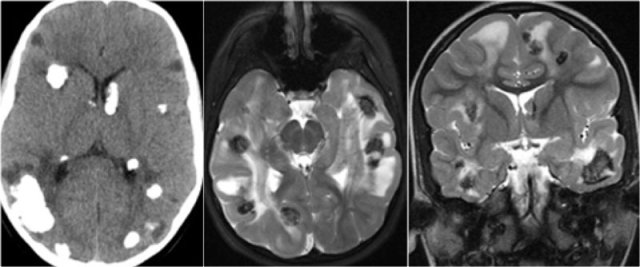
Subependymal giant cell astrocytoma (SEGA)
This is a tumor that develops from a subependymal nodule near the foramen of Monro.
They have a poor prognosis because they lead to obstruction of CSF flow.
They are characterized by marked enhancement and their typical location.
Axial T2WI and T1WI-CE show a giant cell astrocytoma at the level of the left foramen of Monro causing obstructive hydrocephalus.
Also notice tuber on the left.
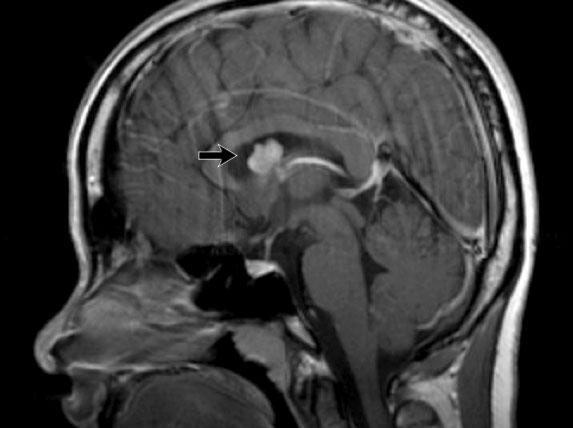
Sagittal T1WI post contrast shows a giant cell astrocytoma in the right foramen of Monro.
Sturge-Weber Syndrome
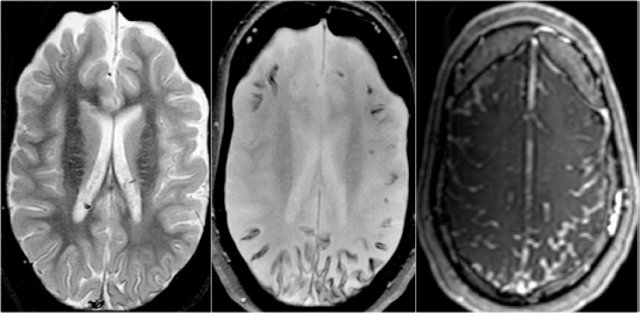 Sturge-Weber. T2WI, SWI and CE-T1WI
Sturge-Weber. T2WI, SWI and CE-T1WI
key findings
- Leptomeningeal enhancement
- Cortical tram-track calcifications
- Atrophy mainly posteriorly
Sturge-Weber is also called encephalotrigeminal angiomatosis.
It is a vascular malformation with capillary venous angiomas in the face (port-wine stain), choroid of the eye and leptomeninges.
Venous occlusion and ischemia lead to angiomatosis with cortical calcium deposition and atrophy
Clinical features are seizures, hemiparesis, anopsia, mental retardation and port-wine stain.
The MR-images show leptomeningeal angiomatosis which is mainly localized in the occipital lobes.
Venous stasis and calcifications are best seen on the SWI.
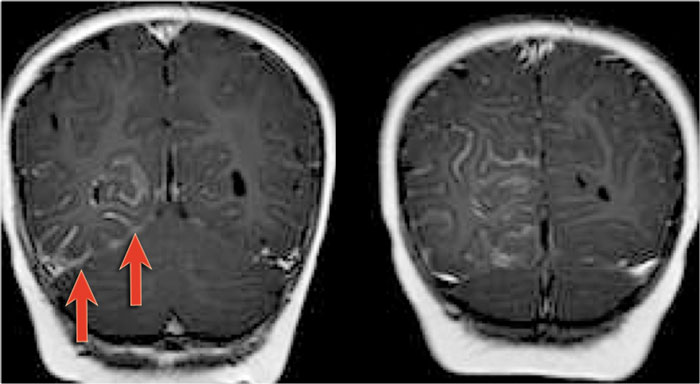 Sturge-Weber angiomatosis. CE-T1WI. Courtesy of Alessandra D' Amico
Sturge-Weber angiomatosis. CE-T1WI. Courtesy of Alessandra D' Amico
MRI in patients with Sturge-Weber can show:
- Atrophy
- High signal on T2WI due to gliosis
- Low signal in areas with calcifications.
- Leptomeningeal enhancement
Coronal MR-images of a patient with Sturge-Weber show leptomeningeal enhancement in the right posterior hemispere.
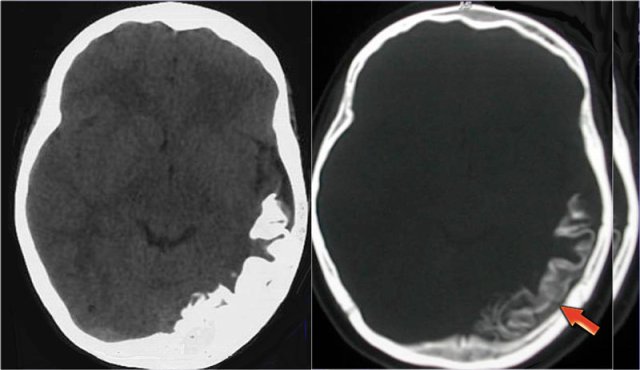 Sturge-Weber angiomatosis. Courtesy of Alessandra D' Amico
Sturge-Weber angiomatosis. Courtesy of Alessandra D' Amico
CT in a patient with Sturge-Weber shows huge cortical and subcortical tram-track calcifications involving the left posterior hemispere.
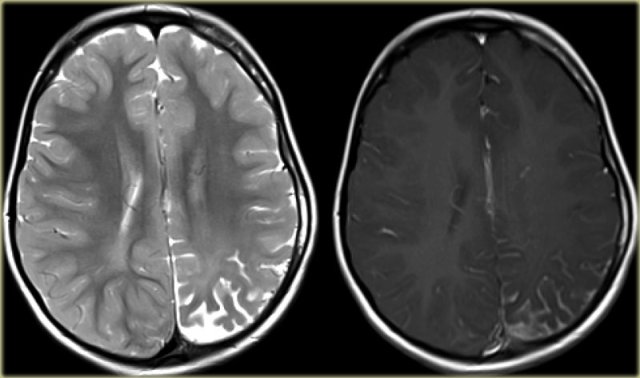 Sturge-Weber. T2WI and CE-T1WI
Sturge-Weber. T2WI and CE-T1WI
4-year-old boy with Sturge-Weber syndrome.
Notice atrophy of the left posterior cerebral hemisphere with leptomeningeal enhancement and thickening.
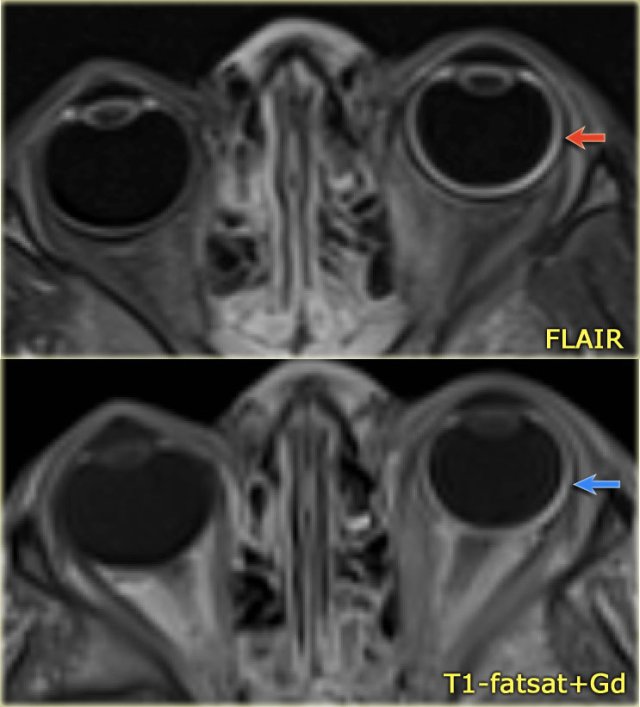 Sturge-Weber. Axial FLAIR and CE-T1WI with fatsat. Diffuse choroidal hemangioma
Sturge-Weber. Axial FLAIR and CE-T1WI with fatsat. Diffuse choroidal hemangioma
Diffuse choroidal hemangioma
In Sturge-Weber a vascular malformation of the choroid of the eye is seen.
These patients present with buphthalmos (enlarged eye) due to increased intraocular pressure and hemianopsia.
Eye abnormalities in a 4-year-old boy with Sturge-Weber syndrome.
Notice FLAIR-hyperintensity (red arrow) and excessive enhancement of the wall of the left globe (blue arrow) consistent with a diffuse choroidal hemangioma.
Polymicrogyria
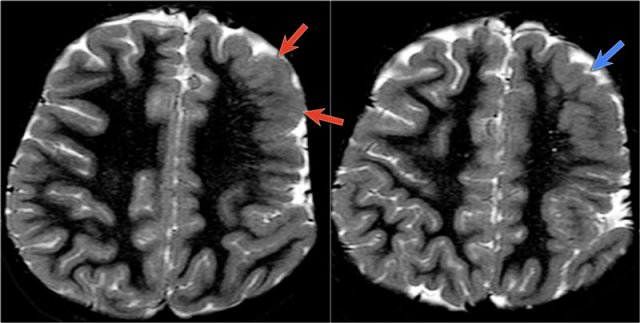
key findings
- Numerous small gyri
- Predilection for Sylvian fissure
- Atrophy mainly posteriorly
- Anomalous venous drainage in areas of polymicrogyria
Polymicrogyria is a malformation due to an alteration of the cortical development in the late stage of neuronal migration.
The deeper layers of the cortex form multiple small gyri with derangement of the normal lamination and sulcation.
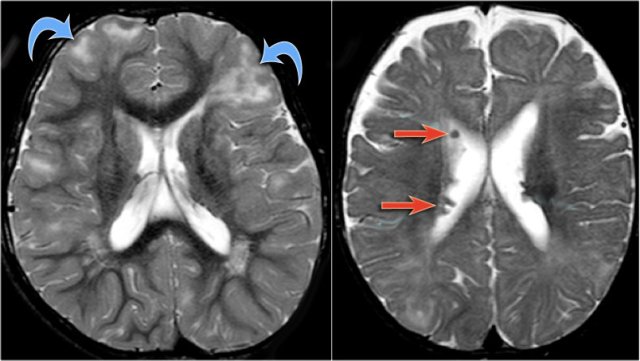
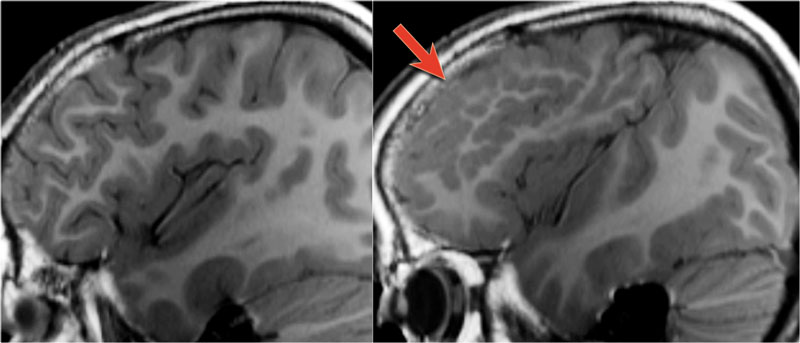 LEFT: normal RIGHT: polymicrogyria (arrow)
LEFT: normal RIGHT: polymicrogyria (arrow)
The T1W-images show a comparison between normal lamination and sulcation on the left and polymicrogyria on the right (arrow).
Heterotopia
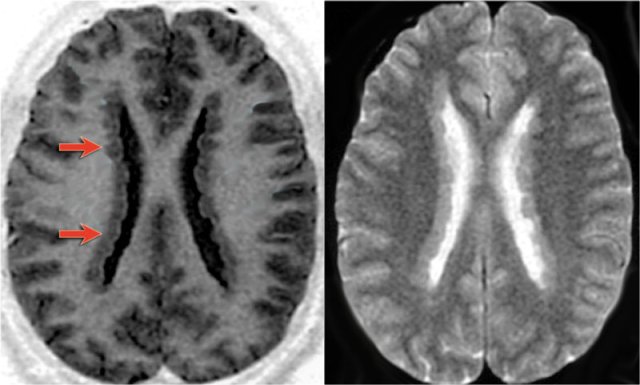 Heterotopia: subependymal nodules (arrows).
Heterotopia: subependymal nodules (arrows).
Heterotopic Grey Matter results from an arrested migration of normal neurons along the radial path between the ventricular walls (ependyma) and the subcortical regions.
There are two types of heterotopia: subependymal and subcortical.
The most common clinical presentation is intractable seizures.
Heterotopia present as nodular foci of grey matter intensity on all sequences. They do not enhance.
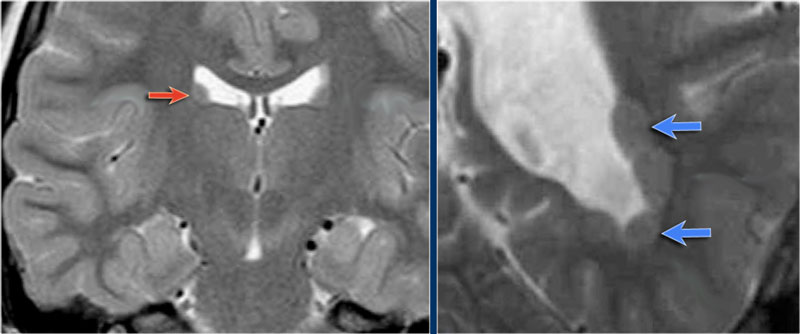 Heterotopia
Heterotopia
Images of a typical subependymal heterotopia.
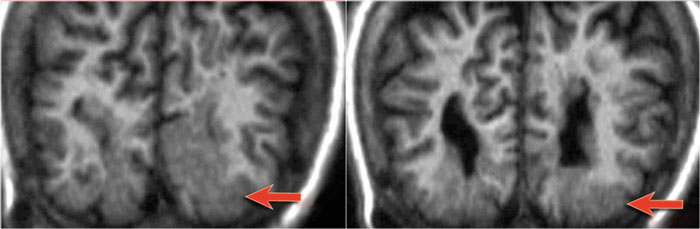 Courtesy of Alessandra D' Amico
Courtesy of Alessandra D' Amico
Another case of heterotopia with typical subcortical nodules (arrows).
Schizencephaly
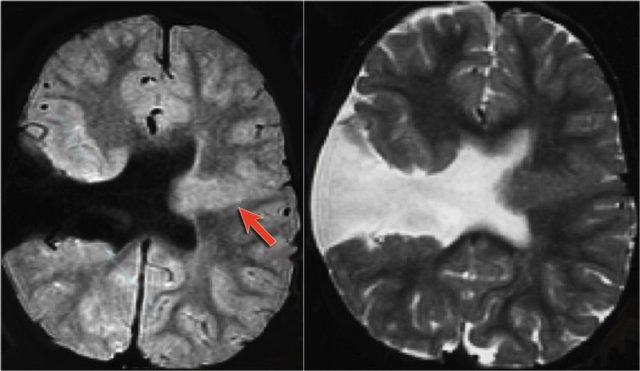 Schizencephaly
Schizencephaly
Schizencephaly is a cleft in the brain that connects the lateral ventricle to the subarachnoid space.
The cleft is lined by polymicrogyric gray matter.
Open-lip schizencephaly is characterized by separation of the cleft walls.
Closed-lip schizencephaly is characterized by cleft walls in apposition to each other.
Patients have seizures and hemiparesis, which is proportional to the size of the cleft and are more common in the open-lip type.
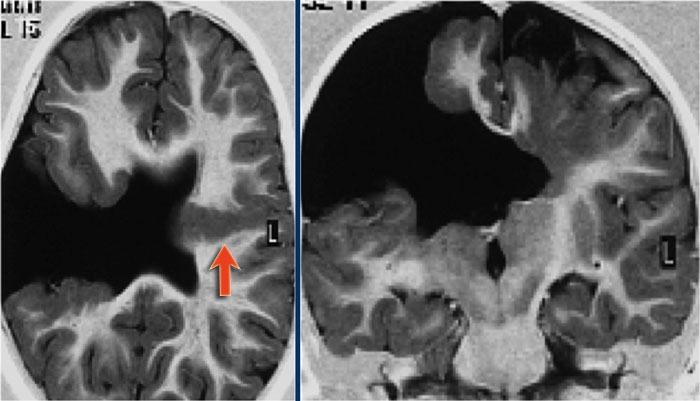 Schizencephaly. Courtesy of Alessandra D' Amico
Schizencephaly. Courtesy of Alessandra D' Amico
First study the images and then continue reading.
This patient has a bilateral schizencephaly.
There is an open-lip type on the right and a closed-lip type on the left (red arrow).
Notice the track of grey matter in the left hemisphere on the axial image.
The differential diagnosis of schizencaphaly is porencephaly, which is also a cleft, but it is not lined by grey matter.

Charity
All the profits of the Radiology Assistant go to Medical Action Myanmar which is run by Dr. Nini Tun and Dr. Frank Smithuis sr, who is a professor at Oxford university and happens to be the brother of Robin Smithuis.
Click here to watch the video of Medical Action Myanmar and if you like the Radiology Assistant, please support Medical Action Myanmar with a small gift